神奈川県保険医協会とは
開業医を中心とする保険医の生活と権利を守り、
国民の健康と医療の向上を目指す
TOP > 神奈川県保険医協会とは > 私たちの考え > 2019/10/28 政策部長談話 「第Ⅲ相治験を省略した医薬品の製造販売の法制化 薬機法改正法案の拙速を警鐘する」
2019/10/28 政策部長談話 「第Ⅲ相治験を省略した医薬品の製造販売の法制化 薬機法改正法案の拙速を警鐘する」
第Ⅲ相治験を省略した医薬品の製造販売の法制化
薬機法改正法案の拙速を警鐘する
神奈川県保険医協会
政策部長 桑島 政臣
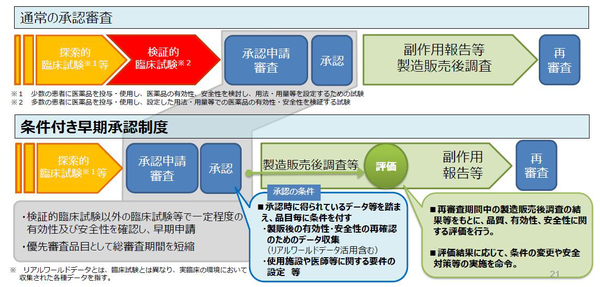
医薬品医療機器法(薬機法)の改正案が、この臨時国会で審議される。通常国会からの継続審査であり、本格審議は11月からと目されている。法案はいくつか問題を含むが、医薬品の製造販売の承認にあたり、治験の第Ⅲ相試験(検証的臨床試験)の省略を可とする、「条件付き早期承認」の法制化が盛り込まれている。諸外国にはないこの仕組みの法制化は慎重さが必要と考える。われわれは、そのことを警鐘する。
◆ 成長戦略と軌を一にした「条件付き早期承認」
医薬品の開発、製造・販売は、①「くすりの候補」(医薬品のモトとなる新規物質)の合成・発見の「基礎研究」、②新規物質の有効性と安全性を研究(動物が対象)する「非臨床試験」、③人を対象とした有効性と安全性のテストとなる「治験(臨床研究)」、④製薬企業から厚労省への「承認申請」とPMDA ((独)医薬品医療機器総合機構)による「審査」、⑤薬事承認、⑥薬価収載・製造販売、の順を経ていき、⑦市販後調査がなされる。その中の「治験」は、第Ⅰ相(比較的少数の健康な人が対象)、第Ⅱ相(少数の患者が対象)、第Ⅲ相(多数の患者が対象)と、順に進んでいく。因みに第Ⅱ相試験は「少数の患者」に医薬品を投与し、医薬品の有効性、安全性を「検討」し、用法・用量等を「設定」するための試験、第Ⅲ相試験は「多数の患者」に医薬品を投与し、設定した用法・用量等で、医薬品の有効性・安全性を「検証」する試験である。
よって医薬品の製造販売の承認(薬事承認)に関し、第Ⅲ相臨床試験を経て製造企業が申請するのが通常である。この通常の承認審査を緩め、製造販売後の調査実施を条件に付与し探索的臨床試験(第Ⅱ相試験)のみで承認し、製品化・販売を認めるのが「条件付き早期承認制度」である。これは成長戦略の一環としてH29年10月20日の通知で制度化されている。従来、薬機法79条により個別に審査当局との合意で対応されていたものを整理・明確化し、製薬企業の開発予見性を高め、早期実用化を推進するとしたのである。
対象の医薬品は、①適応疾患が重篤(致死的疾患、不可逆的進行で日常生活に著しい影響、その他)、②医療上の高い有用性(既存の治療法・予防法等がない等)、③第Ⅲ相試験が困難か、患者少数などで長期間を要す、④第Ⅲ相試験以外の臨床試験等により一定の有効性、安全性が確認できる、の①~④のいずれにも該当する医薬品となっている。この通知運用を、この国会で法文化しより明確化を図るとされ、製薬企業は制度の恒久化と解している。鈴木康裕医務技監は、莫大な費用がかかる第Ⅲ相試験の開発コストを低減し、薬価を抑えながら早期アクセスを確保する、実臨床で第Ⅲ相試験を行う制度だと、その本音を先頃、吐露している。
◆ 先行する再生医療品の「条件及び期限付き承認」の舞台裏
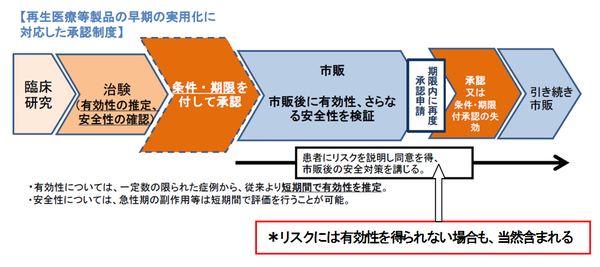
この「条件付き早期承認」に先立ち、「条件及び期限付き承認」の仕組みが、2013年(H25年)の薬事法改正で、再生医療等製品を法的に位置づけるとともにそれを対象に法文化、制度化が先行してなされている。この再生医療等製品は、治験での安全性・有効性の確認に関し、有効性は「推定」段階で製品化・販売を認め、市販後7年以内の期限で有効性と更なる安全性を検証し、再度、正式に承認申請し審査後、本承認とするものである。有効性の「確定」と違い、数千例の症例データは不要で、数十例の症例データで製品化が可能となっている。現在、この条件付き期限付き承認の再生医療等製品は3品目ある。
この法制化の背景には再生医療の重点化が2013年の日本再興戦略に位置づき、世界一迅速に製品化、「実用化」ができるように法改定が行われたということがある。しかも、旗振りの規制改革会議の委員には、再生医療製品の企業の創業者が当時より就いている。最近、その該当企業より、日本初の遺伝子治療薬が条件付き期限付き承認で製品化が図られたが、薬価が想定より低かったため株価がストップ安になったことが新聞で話題となっている。医薬品の高薬価へ投機筋が食指を伸ばす構図が顕在化している。
なお、同時期にこれを後押しする形で、医療保険のサイドで、患者申出療養が法定化されて制度化となっている。これは臨床研究の実施計画が成立しないものや、実施計画の被験者(患者)の適格基準に外れ登録できなかった方への、臨床研究からも外れた未確立技術や未承認医薬品の実施・投与と、医療保険との混合診療(保険外併用療養)である。更には、「拡大治験」が制度化され、治験に適格基準外で登録できない被験者(患者)への治験薬使用が認められ(実薬単群非盲検試験)、評価療養として混合診療(保険外併用療養)の対象とされている。いわば医薬品の承認、臨床研究、保険診療の各制度の融解と混然一体化の進展である。
◆ 「条件付き早期承認」と再生医療品の「条件付き期限付き承認」の差異
今回の「条件付き早期承認」は、再生医療等製品「以外」を対象に法制化を行うものである。再生医療等製品の「条件及び期限付き承認」と違い、製品化・販売後に「検証」はかけず、有効性や安全性を示す実臨床のリアルデータの収集で可としている。再生医療品の「検証」は、治験を意味しておらず、その方法は「ケースバイ・ケース」(医療機器審査管理課)でとしているが、それよりも緩いものとなっている。
条件付き早期承認は、通知で制度化された以降、肺がんと固形がんの抗がん剤、2品目が認められている。両者の条件は概ね、製造販売後、全症例を対象に使用成績調査の実施、安全性及び有効性に関するデータを早期に収集等としており、あとは通常の医薬品の再評価のレールに乗るだけとなっており、検証措置がない。薬害オンブズパーソン会議は制度停止と法制化へ危惧を示す意見書(2018.5.29)を既に大臣へ提出している。
今回の法制化は、医薬品の承認申請の基準緩和の恒久化であるだけに、今後に禍根を残す。このほか薬機法改定は、審査期間短縮の医薬品の先駆け審査の法制化や、実用実績も検証も乏しいオンライン服薬指導の解禁など問題がある。われわれは薬機法改正の慎重審議、撤回を改めて求める。
2019年10月28日
◆「条件付き早期承認制度」の法制化(医薬品) * 厚労省資料より
<参考>再生医療等製品の「条件及び期限付き承認制度」 * PMDA資料より
第Ⅲ相治験を省略した医薬品の製造販売の法制化
薬機法改正法案の拙速を警鐘する
神奈川県保険医協会
政策部長 桑島 政臣
医薬品医療機器法(薬機法)の改正案が、この臨時国会で審議される。通常国会からの継続審査であり、本格審議は11月からと目されている。法案はいくつか問題を含むが、医薬品の製造販売の承認にあたり、治験の第Ⅲ相試験(検証的臨床試験)の省略を可とする、「条件付き早期承認」の法制化が盛り込まれている。諸外国にはないこの仕組みの法制化は慎重さが必要と考える。われわれは、そのことを警鐘する。
◆ 成長戦略と軌を一にした「条件付き早期承認」
医薬品の開発、製造・販売は、①「くすりの候補」(医薬品のモトとなる新規物質)の合成・発見の「基礎研究」、②新規物質の有効性と安全性を研究(動物が対象)する「非臨床試験」、③人を対象とした有効性と安全性のテストとなる「治験(臨床研究)」、④製薬企業から厚労省への「承認申請」とPMDA ((独)医薬品医療機器総合機構)による「審査」、⑤薬事承認、⑥薬価収載・製造販売、の順を経ていき、⑦市販後調査がなされる。その中の「治験」は、第Ⅰ相(比較的少数の健康な人が対象)、第Ⅱ相(少数の患者が対象)、第Ⅲ相(多数の患者が対象)と、順に進んでいく。因みに第Ⅱ相試験は「少数の患者」に医薬品を投与し、医薬品の有効性、安全性を「検討」し、用法・用量等を「設定」するための試験、第Ⅲ相試験は「多数の患者」に医薬品を投与し、設定した用法・用量等で、医薬品の有効性・安全性を「検証」する試験である。
よって医薬品の製造販売の承認(薬事承認)に関し、第Ⅲ相臨床試験を経て製造企業が申請するのが通常である。この通常の承認審査を緩め、製造販売後の調査実施を条件に付与し探索的臨床試験(第Ⅱ相試験)のみで承認し、製品化・販売を認めるのが「条件付き早期承認制度」である。これは成長戦略の一環としてH29年10月20日の通知で制度化されている。従来、薬機法79条により個別に審査当局との合意で対応されていたものを整理・明確化し、製薬企業の開発予見性を高め、早期実用化を推進するとしたのである。
対象の医薬品は、①適応疾患が重篤(致死的疾患、不可逆的進行で日常生活に著しい影響、その他)、②医療上の高い有用性(既存の治療法・予防法等がない等)、③第Ⅲ相試験が困難か、患者少数などで長期間を要す、④第Ⅲ相試験以外の臨床試験等により一定の有効性、安全性が確認できる、の①~④のいずれにも該当する医薬品となっている。この通知運用を、この国会で法文化しより明確化を図るとされ、製薬企業は制度の恒久化と解している。鈴木康裕医務技監は、莫大な費用がかかる第Ⅲ相試験の開発コストを低減し、薬価を抑えながら早期アクセスを確保する、実臨床で第Ⅲ相試験を行う制度だと、その本音を先頃、吐露している。
◆ 先行する再生医療品の「条件及び期限付き承認」の舞台裏
この「条件付き早期承認」に先立ち、「条件及び期限付き承認」の仕組みが、2013年(H25年)の薬事法改正で、再生医療等製品を法的に位置づけるとともにそれを対象に法文化、制度化が先行してなされている。この再生医療等製品は、治験での安全性・有効性の確認に関し、有効性は「推定」段階で製品化・販売を認め、市販後7年以内の期限で有効性と更なる安全性を検証し、再度、正式に承認申請し審査後、本承認とするものである。有効性の「確定」と違い、数千例の症例データは不要で、数十例の症例データで製品化が可能となっている。現在、この条件付き期限付き承認の再生医療等製品は3品目ある。
この法制化の背景には再生医療の重点化が2013年の日本再興戦略に位置づき、世界一迅速に製品化、「実用化」ができるように法改定が行われたということがある。しかも、旗振りの規制改革会議の委員には、再生医療製品の企業の創業者が当時より就いている。最近、その該当企業より、日本初の遺伝子治療薬が条件付き期限付き承認で製品化が図られたが、薬価が想定より低かったため株価がストップ安になったことが新聞で話題となっている。医薬品の高薬価へ投機筋が食指を伸ばす構図が顕在化している。
なお、同時期にこれを後押しする形で、医療保険のサイドで、患者申出療養が法定化されて制度化となっている。これは臨床研究の実施計画が成立しないものや、実施計画の被験者(患者)の適格基準に外れ登録できなかった方への、臨床研究からも外れた未確立技術や未承認医薬品の実施・投与と、医療保険との混合診療(保険外併用療養)である。更には、「拡大治験」が制度化され、治験に適格基準外で登録できない被験者(患者)への治験薬使用が認められ(実薬単群非盲検試験)、評価療養として混合診療(保険外併用療養)の対象とされている。いわば医薬品の承認、臨床研究、保険診療の各制度の融解と混然一体化の進展である。
◆ 「条件付き早期承認」と再生医療品の「条件付き期限付き承認」の差異
今回の「条件付き早期承認」は、再生医療等製品「以外」を対象に法制化を行うものである。再生医療等製品の「条件及び期限付き承認」と違い、製品化・販売後に「検証」はかけず、有効性や安全性を示す実臨床のリアルデータの収集で可としている。再生医療品の「検証」は、治験を意味しておらず、その方法は「ケースバイ・ケース」(医療機器審査管理課)でとしているが、それよりも緩いものとなっている。
条件付き早期承認は、通知で制度化された以降、肺がんと固形がんの抗がん剤、2品目が認められている。両者の条件は概ね、製造販売後、全症例を対象に使用成績調査の実施、安全性及び有効性に関するデータを早期に収集等としており、あとは通常の医薬品の再評価のレールに乗るだけとなっており、検証措置がない。薬害オンブズパーソン会議は制度停止と法制化へ危惧を示す意見書(2018.5.29)を既に大臣へ提出している。
今回の法制化は、医薬品の承認申請の基準緩和の恒久化であるだけに、今後に禍根を残す。このほか薬機法改定は、審査期間短縮の医薬品の先駆け審査の法制化や、実用実績も検証も乏しいオンライン服薬指導の解禁など問題がある。われわれは薬機法改正の慎重審議、撤回を改めて求める。
2019年10月28日
◆「条件付き早期承認制度」の法制化(医薬品) * 厚労省資料より
<参考>再生医療等製品の「条件及び期限付き承認制度」 * PMDA資料より